China: Regulatory updates on product-related license for API
Written By

Partner
China
I am a partner in the international Corporate Group based in Shanghai and have been living and working in China since 1999, and based in Shanghai since 2003. I have close to 20 years' experience practising law in China.
Related
Practices
Sectors
Regions
Countries
Life Sciences & Healthcare Newsletter: Hong Kong & China Update August 2018
While more work needs to be done to bring the Chinese regulatory landscape up to the standards of other industry leading countries, as exemplified in the recent incidents concerning vaccines in the country, China has just made a great step in the direction of global pharmaceutical regulatory standards with the introduction of a Drug Master File ("DMF") regime.
In order to accelerate the evaluation procedure for market authorizations of new drugs, the Opinions on Deepening the Reform of the Evaluation and Approval System and Inspiring Innovation of Drugs and Medical Devices issued by State Council on 8 October 2017 have introduced a DMF like joint assessment, which integrates the evaluation and approval of active pharmaceutical ingredients (API), excipients and packaging materials with the finished drugs.
1. DMF Code
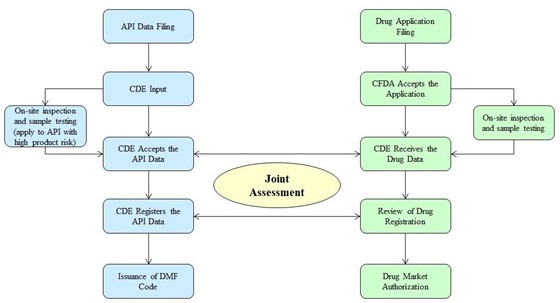
Based on the Opinions, the CFDA has issued the No. [2017] 146 Announcement on Adjusting the Evaluation and Approval of API, Excipients and Packaging Materials on 30 November 2017. In the past, the manufacturing and distribution of API was regulated in the same way as for the finished drug, which requires the product related approval i.e. the drug approval numbers.
However, from 30 November 2017, the CFDA will not accept any new applications or issuing new drug approval numbers to register an API alone to be used in any drug under the new version of the Chemical Drug Classifications 2.2, 2.3 2.4, 3, 4, 5. The manufacturer or importer of above API shall submit the API registration material to the database of the Centre for Drug Evaluation ("CDE") to obtain a DMF code instead. The information registered to obtain the DMF code will include
(i) a basis description,
(ii) manufacturing information,
(iii) special nature evaluation,
(iv) quality control procedures,
(v) comparison of product,
(vi) packaging material and stability, amongst others, and all in accordance with the CFDA No. [2016] 80 Notice on Chemical Drug Classification and Application Material Requirement.
The DMF code holder will be required to submit a quality management report every year.
For the upstream market of API, the finished drug registration applicant will be responsible for the overall quality of the drugs and shall fully evaluate the impact on the drug quality from API, excipients and packaging materials.
2. Import of API
CFDA has also issued the No. [2018] 8 Notice on Matters regarding Import of API, Excipients and Packaging Materials1 in April 2018 to clarify the requirements in order to get clearance of imported API. In the past, the import of API required, inter alia, the Imported Drug Licenses (IDLs) for the issuance of Imported Drug Customs Clearance Letter which is an equivalent procedure as for finished drugs.
Upon the Announcement of No. [2016] 80, if an importer has obtained the IDL before the promulgation of No. [2017] 146, customs authorities shall still issue the Imported Drug Customs Clearance Letter for the clearance of imported API in the following situations:
- The IDL is still within its valid period. If the IDL expires, such API may be imported for the same previous use or for study purposes only;
- The DMF code has been obtained and approved to be used in the drug with market authorization; such API can be imported for the use in the approved finished drug or for study purposes;
- The DMF code has been obtained but not approved to be used in any drug with market authorization yet. Such API may be imported for study purposes only;
- The DMF code has not been obtained but the Imported Drug Approval has been issued for study purposes only; or
- Other approvals issued for the import of the specific API.
3. Implication
Under the new regime, applicants of new drug registrations or any change registration can only choose the API with DMF code for study and registration except for API manufactured for self-use or supplied to specific Market Authorization Holders. As the responsible party for the drug quality, the drug applicant will bear the product liability towards consumers and the regulatory authorities but shall be indemnified contractually by API suppliers for any quality defects of the API product. This will require the drug manufacturers to pay specific attention to this point when negotiating their supply contracts in order to enhance management of this particular liability risk.
Holders of Drug Approval Numbers and IDLs shall start to prepare the registration of DMF codes if there is a business plan to supply the products to new customers or for different finished drugs in place. DMF holders will have more direct statutory obligations towards the drug applicants, such as to make notifications of any change or defect in the respective API product. The DMF data will not be disclosed to the drug applicants, and therefore the confidentiality of the API data and the core know-how from the drug registration applicant I supposed to be ensured.
For the regulators, the drug registration applicant can link their product applications as below with the API data under the DMF code if the DMF code holder issues an authorization letter to authorize the CFDA to access API data. Such API data will only be assessed and examined by the CDE in conjunction with the drug application as a whole master file. This will prevent that the CDE faces unnecessary workload due to the need to review applications of API not being used in any finished drug.



{kind=link}